|
|
CURSO DE BIOLOGÍAAlejandro Porto Andión |
|
|
|
|
|
|
|
Inicio Temas de Células Aula virtual |
|
|
|
|
![]()
TEMA 14: ENZIMAS.
Los enzimas son biomoléculas especializadas en la catálisis de las reacciones químicas que tienen lugar en la célula. Son muy eficaces como catalizadores ya que son capaces de aumentar la velocidad de las reacciones químicas mucho más que cualquier catalizador artificial conocido, y además son altamente específicos ya que cada uno de ellos induce la transformación de un sólo tipo de sustancia y no de otras que se puedan encontrar en el medio de reacción.
1.-CATÁLISIS QUÍMICA.
Antes de comenzar a analizar la naturaleza y función de los enzimas, resulta conveniente enunciar algunos conceptos básicos acerca de la velocidad de las reacciones químicas y el fenómeno de la catálisis.
La teoría cinética química establece que las reacciones químicas transcurren molécula a molécula de modo que una reacción tal como
R (reactivos) —› P (productos)
tiene lugar porque una determinada fracción de la población de moléculas R, en un instante dado, posee energía suficiente como para alcanzar un estado activado llamado estado de transición, en el que es muy fácil que se rompan o se formen uno o más enlaces químicos para formar los productos P. Es frecuente confundir el estado de transición con un intermediario de reacción; sin embargo este estado no es ninguna especie química concreta, sino que podría definirse con más exactitud como un "momento molecular fugaz", altamente inestable, en el que uno o más enlaces químicos están muy próximos a romperse o a formarse.
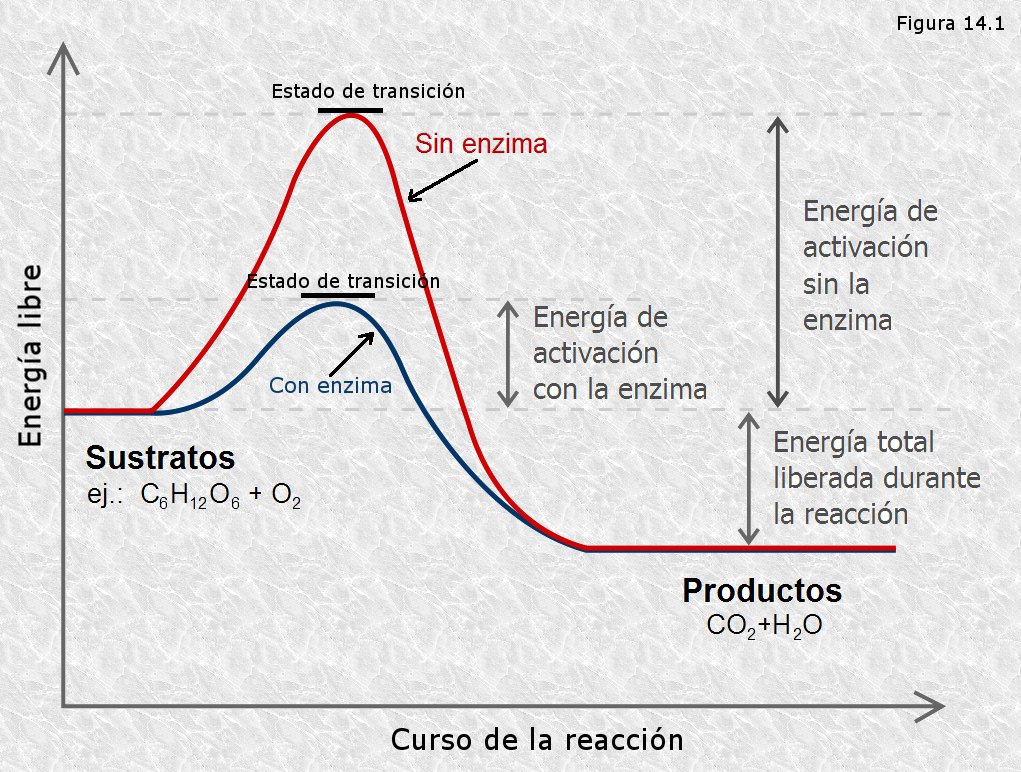
La velocidad de una reacción química es proporcional al número de moléculas por unidad de tiempo con energía suficiente para alcanzar el estado de transición. Este estado de transición posee una energía superior a la de los reactivos y a la de los productos constituyendo entre ellos una barrera energética que debe superarse para que la reacción tenga lugar. La diferencia entre la energía de los reactivos y la del estado de transición recibe el nombre de energía libre de activación (ver Figura 14.1).
Existen dos métodos generales mediante los cuales puede acelerarse la velocidad de una reacción química. Uno de ellos consiste en la elevación de la temperatura de modo que al incrementarse el movimiento térmico de las moléculas reaccionantes aumenta la fracción de moléculas que poseen energía suficiente para alcanzar el estado de transición. El otro método consiste en usar un catalizador, sustancia que se combina de un modo transitorio con los reaccionantes de manera que éstos alcanzan un estado de transición de menor energía de activación; cuando se forman los productos se regenera el catalizador libre. Así, un catalizador es una sustancia que, sin consumirse en el proceso, aumenta la velocidad de una reacción química rebajando la barrera de energía de activación (ver Figuras 14.1). Es conveniente resaltar el hecho de que los catalizadores no alteran los equilibrios de las reacciones químicas, sólo consiguen que dichos equilibrios se alcancen más rápidamente de lo que lo harían en ausencia de catalizador.
2.-INTRODUCCIÓN AL ESTUDIO DE LOS ENZIMAS.
Gran parte de la historia de la Bioquímica discurre pareja a la historia de la investigación de los enzimas. Citaremos algunos hitos importantes:
-La existencia de catalizadores biológicos fue descrita por primera vez a comienzos del S. XIX en estudios acerca de la digestión de la carne por secreciones del estómago y la conversión del almidón en azúcar por la saliva.
-En 1835 la primera teoría general sobre la catálisis química publicada por J.J. Berzelius incluía un ejemplo de lo que hoy conocemos como un enzima: la diastasa de la malta, señalando que la hidrólisis del almidón se catalizaba más eficazmente por ésta que por el ácido sulfúrico.
-En 1860 Louis Pasteur propuso que la fermentación del azúcar para transformarse en alcohol era inducida por ciertos catalizadores biológicos, razón por la cual los enzimas fueron llamados inicialmente "fermentos". Pasteur supuso que dichos catalizadores se hallaban unidos de modo indisoluble a la estructura de las células de la levadura por lo que no podían actuar fuera de estas.
-En 1877 se utiliza por primera vez la denominación "enzima" (etimológicamente "en la levadura").
-En 1897 E. Büchner consiguió extraer de las células de la levadura los enzimas que catalizan la fermentación alcohólica, demostrando que éstos pueden actuar independientemente de la estructura celular. Este hecho permitió estudiar "in vitro" la actividad y propiedades de los enzimas, aislarlos en estado puro y analizar su composición.
-En 1926 J.B. Sumner aisló un enzima, la ureasa, en forma cristalina pura y demostró que los cristales estaban formados por proteínas.
-Entre 1930 y 1936 se aislaron en forma cristalina pura diversos enzimas quedando establecida de modo definitivo la naturaleza proteica de estos catalizadores biológicos. Desde entonces se han identificado varios miles de enzimas diferentes, habiéndose aislado muchos de ellos en forma cristalina.
-En el mismo período J.B.S. Haldane expuso la idea de que los enzimas establecen interacciones débiles con el sustrato para distorsionarlo y catalizar así su transformación; esta idea resultó capital para el moderno conocimiento de la acción enzimática.
-En 1981 se descubrió que determinados tipos de RNA pueden catalizar su propia síntesis, hecho este que obligó a revisar algunas de las ideas preexistentes acerca de la naturaleza de los biocatalizadores.
En la actualidad se considera que, con la excepción de un reducido grupo de moléculas de RNA con propiedades catalíticas, los enzimas son proteínas, y como tales, exhiben todas las propiedades inherentes a este grupo de biomoléculas. Los pesos moleculares de las proteínas enzimáticas oscilan desde unos 12.000 daltons hasta más de un millón.
En muchos casos, las cadenas polipeptídicas de la proteína enzimática son suficientes para que ésta desarrolle su actividad catalítica. En otros, se hace necesaria la participación de un compuesto químico adicional, de naturaleza no proteica, denominado cofactor.
3.-ESTRUCTURA DE LOS ENZIMAS: EL CENTRO ACTIVO.
Los enzimas, en cuanto que proteínas, presentan todos los rasgos estructurales y propiedades químicas que caracterizan a esta notable clase de biomoléculas. En efecto, se ha podido comprobar que los enzimas pierden su actividad catalítica cuando sufren desnaturalización por efecto de los mismos agentes que afectan a las demás proteínas; la conformación tridimensional nativa intacta de la proteína enzimática resulta indispensable para que ésta desempeñe su función. Además, los enzimas, al igual que otras muchas clases de proteínas, presentan un centro activo a través del cual interactúan con la(s) molécula(s) de ligando, que en este caso recibe(n) el nombre de sustrato(s), mediante un acoplamiento espacial (las superficies moleculares de ambos tienen formas complementarias) y químico (grupos funcionales complementarios del enzima y el (los) sustrato(s) establecen diferentes tipos de interacciones débiles entre sí). Tanto la actividad catalítica como el elevado grado de especificidad química que presentan los enzimas residen en esta interacción específica entre el enzima y su sustrato.
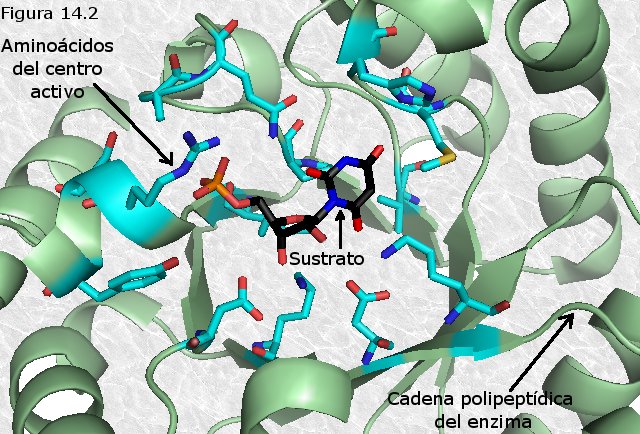 El centro activo es una cavidad existente en la
superficie del enzima que está forrada interiormente por una serie de
restos de aminoácidos (Figura
14.2). Por regla general los aminoácidos que forman parte del centro
activo no se encuentran contiguos en la cadena polipeptídica, sino
ocupando posiciones a veces muy alejadas en la misma. El hecho de que
estos aminoácidos coincidan próximos entre sí sobre el centro activo no
es más que una consecuencia del plegamiento característico de la cadena
polipeptídica, es decir, de la conformación tridimensional nativa de la
proteína enzimática. Puede resultar útil, aunque no siempre posible,
distinguir entre los aminoácidos que forman parte del centro activo dos
categorías:
El centro activo es una cavidad existente en la
superficie del enzima que está forrada interiormente por una serie de
restos de aminoácidos (Figura
14.2). Por regla general los aminoácidos que forman parte del centro
activo no se encuentran contiguos en la cadena polipeptídica, sino
ocupando posiciones a veces muy alejadas en la misma. El hecho de que
estos aminoácidos coincidan próximos entre sí sobre el centro activo no
es más que una consecuencia del plegamiento característico de la cadena
polipeptídica, es decir, de la conformación tridimensional nativa de la
proteína enzimática. Puede resultar útil, aunque no siempre posible,
distinguir entre los aminoácidos que forman parte del centro activo dos
categorías:
a)Aminoácidos catalíticos.- Son uno o más aminoácidos cuyas cadenas laterales R poseen unas peculiaridades químicas tales que los facultan para desarrollar una función catalítica. Constituyen el verdadero centro catalítico del enzima.
b)Aminoácidos de unión.- Son una serie de aminoácidos cuyas cadenas laterales R poseen grupos funcionales que pueden establecer interacciones débiles (puentes de hidrógeno, interacciones iónicas, etc.) con grupos funcionales complementarios de la molécula de sustrato. Su función consiste en fijar la molécula de sustrato al centro activo en la posición adecuada para que los aminoácidos catalíticos puedan actuar (ver Figura 14.2).
En cuanto al resto de los aminoácidos de la cadena polipeptídica del enzima, los que no forman parte del centro activo, podría pensarse en principio que no desempeñan ninguna función, pero esto no es cierto; tienen la importante misión de mantener la conformación tridimensional catalíticamente activa del enzima; sin ella no existiría centro activo y el enzima no podría interactuar con su sustrato.
4.-CINÉTICA ENZIMÁTICA: EL COMPLEJO ENZIMA-SUSTRATO.
Los enzimas actúan de acuerdo con los mismos principios generales que los demás catalizadores: aumentan la velocidad de las reacciones químicas combinándose transitoriamente con los reactivos de manera que estos alcanzan un estado de transición con una energía de activación menor que el de la reacción no catalizada. Hay que destacar sin embargo que los enzimas son mucho más eficaces que cualquier catalizador artificial conocido. Se ha podido comprobar que los aumentos de velocidad que producen son de entre 107 y 1014 veces la velocidad de la reacción no catalizada. La actividad molecular (número de moléculas de sustrato transformadas por una sola molécula de enzima por minuto) de distintos enzimas oscila entre unos pocos miles y varios millones de moléculas de sustrato por minuto. Cabe preguntarse pues, cómo consiguen los enzimas aumentos tan espectaculares en la velocidad de las reacciones químicas que catalizan.
 Los métodos experimentales para conocer la actividad
catalítica de los enzimas son cada vez más complejos y sofisticados. Sin
embargo, el método más antiguo utilizado en enzimología, desarrollado
por Leonor Michaelis y Maud Menten (Figura
14.3), y que en la actualidad sigue siendo de gran utilidad,
consiste en analizar como varía la velocidad de las reacciones
catalizadas enzimáticamente en función de algunos parámetros
experimentales como la concentración del sustrato o la del propio
enzima, lo que globalmente se conoce con el nombre de cinética
enzimática.
Los métodos experimentales para conocer la actividad
catalítica de los enzimas son cada vez más complejos y sofisticados. Sin
embargo, el método más antiguo utilizado en enzimología, desarrollado
por Leonor Michaelis y Maud Menten (Figura
14.3), y que en la actualidad sigue siendo de gran utilidad,
consiste en analizar como varía la velocidad de las reacciones
catalizadas enzimáticamente en función de algunos parámetros
experimentales como la concentración del sustrato o la del propio
enzima, lo que globalmente se conoce con el nombre de cinética
enzimática.
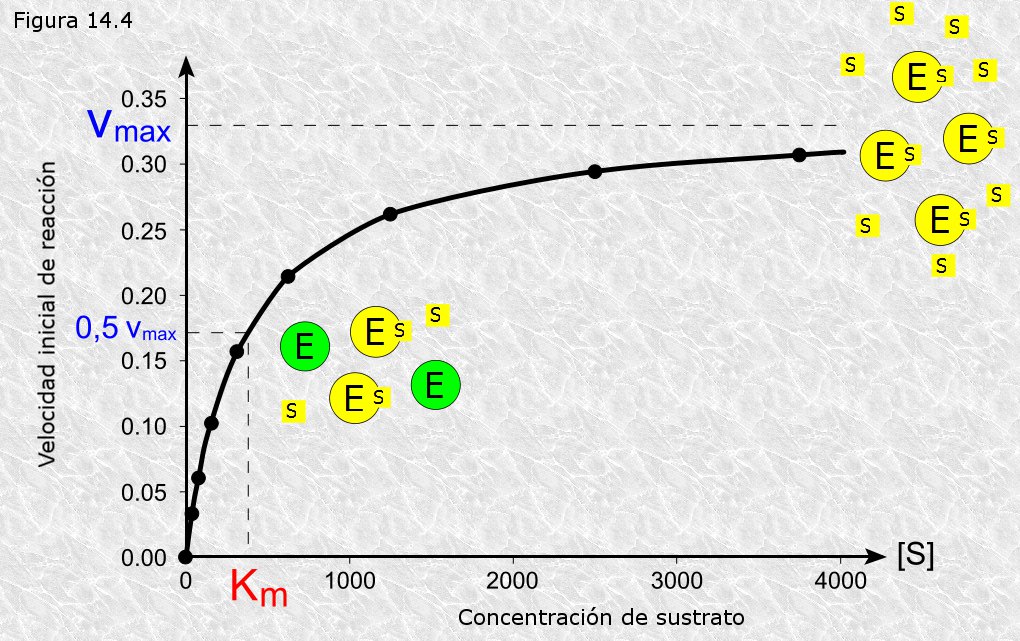
La cinética de las reacciones catalizadas por enzimas muestra un rasgo característico que no se observa en las reacciones no enzimáticas: la la saturación del enzima por el sustrato (ver Figura 14.4). Cuando se mide la velocidad inicial de una reacción catalizada enzimáticamente se observa que para concentraciones de sustrato bajas la velocidad de reacción es proporcional a dicha concentración, como ocurre con carácter general para las reacciones no enzimáticas. A medida que la concentración de sustrato aumenta la velocidad de reacción deja de ser proporcional a ésta. Con un aumento posterior la velocidad de reacción llega a ser totalmente independiente de la concentración del sustrato y se aproxima asimptóticamente a un valor máximo que es característico de cada enzima y que se conoce como velocidad máxima. Se dice entonces que el enzima se halla saturado por el sustrato. La concentración de sustrato a la cual la reacción alcanza la mitad de su velocidad máxima se conoce con el nombre de KM (constante de Michaelis-Menten). KM es un valor característico de cada enzima y constituye una medida de la afinidad del enzima por el sustrato: valores bajos de KM indican una alta afinidad mientras que valores altos representan una baja afinidad.
El efecto de saturación del enzima por el sustrato condujo a los primeros investigadores de la cinética enzimática, incluso antes de que se conociera la naturaleza proteica de los enzimas, a formular la hipótesis, hoy corroborada, de que el enzima y el sustrato se combinan de modo transitorio para formar un complejo enzima-sustrato (Figura 14.5) en el que se alcanza el estado de transición con mayor probabilidad que en la reacción no catalizada. Una vez alcanzado dicho estado el complejo enzima-sustrato se descompone para dar lugar a los productos y el enzima libre.
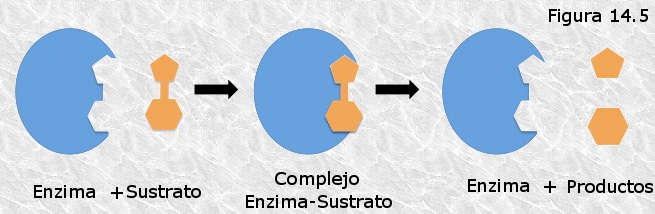
El enzima, una vez liberado, puede combinarse con una nueva molécula de sustrato para formar un nuevo complejo enzima-sustrato cerrándose así el ciclo catalítico del enzima. De este modo, una sola molécula de enzima puede transformar en producto, en sucesivos ciclos catalíticos, a un elevado número de moléculas de sustrato, lo que contribuiría a explicar la gran eficacia catalítica que exhiben estas biomoléculas.
La hipótesis del complejo enzima-sustrato explica de modo satisfactorio el efecto de saturación del enzima por el sustrato observado en los estudios de cinética enzimática: cuando la concentración de sustrato es muy superior a la concentración del enzima en el medio de reacción, todos los centros activos de las moléculas de enzima se hallan ocupados en un momento dado por moléculas de sustrato, con lo que aumentos posteriores de la concentración de éste no se traducen en aumentos en la velocidad de reacción.
5.-CATÁLISIS ENZIMÁTICA: MECANISMOS.
La idea de que el enzima se combina transitoriamente con el sustrato para formar un complejo enzima-sustrato en el cual se alcanza más fácilmente el estado de transición de la reacción catalizada es la piedra angular de todas las explicaciones acerca del fenómeno de la catálisis enzimática. Sin embargo, esta idea no termina de explicar en su totalidad dicho fenómeno; ciertamente, responde a la pregunta de ¿qué hacen los enzimas? pero deja sin contestar otra pregunta esencial: ¿cómo lo hacen?
Una forma interesante de abordar este problema consiste en preguntarse de donde procede la energía necesaria para provocar los considerables descensos en la energía de activación asociados con las reacciones químicas enzimáticamente catalizadas. Si nos atenemos a las leyes termodinámicas, la cuantía de tales descensos debe ser energéticamente "pagada" de alguna manera, ya que la energía "no se crea ni se destruye". La respuesta a esta pregunta reside en la propia naturaleza de la interacción entre el enzima y el sustrato y en el concepto de energía de fijación. Como ya se apuntó anteriormente, la unión entre un enzima y su sustrato para formar el complejo enzima-sustrato se ve favorecida por la formación de una serie de interacciones débiles (puentes de hidrógeno, interacciones iónicas, etc) entre grupos funcionales complementarios de ambas moléculas. La formación de cada una de estas interacciones débiles va acompañada por la liberación de una pequeña cantidad de energía libre que contribuye a estabilizar el complejo. La suma de todas estas pequeñas cantidades de energía liberadas por la totalidad de las interacciones débiles formadas representa la energía de fijación. El papel que juega esta energía en la catálisis enzimática es de una importancia extraordinaria: la energía de fijación no se limita a producir una estabilización del complejo enzima-sustrato, sino que constituye la principal fuente de energía libre que los enzimas utilizan para rebajar la barrera de energía de activación y con ello aumentar la velocidad de la reacción catalizada.
Prosiguiendo con nuestro análisis sobre la naturaleza de la actividad catalítica de los enzimas trataremos ahora de averiguar de qué manera utilizan los enzimas la energía de fijación para producir catálisis. En primer lugar, el enzima puede fijar a las moléculas reaccionantes (sustratos) de manera que queden muy próximas entre sí sobre el centro activo y a su vez próximos a eventuales grupos catalíticos del propio enzima. En segundo lugar, el enzima fija a las moléculas reaccionantes de manera que quedan no sólo próximas entre sí, sino orientadas en la relación geométrica más favorable para que la reacción tenga lugar, es decir, con sus grupos funcionales reaccionantes enfrentados. Las colisiones al azar de moléculas reaccionantes en el medio de reacción son extremadamente improbables, y aún en el caso de producirse, la orientación de los grupos funcionales reaccionantes no será la más favorable en la mayor parte de los casos. Sin embargo, el centro activo del enzima, al fijar a los sustratos de manera energéticamente favorable mediante interacciones débiles, y al hacerlo con una orientación precisa, proporciona a éstos un "lugar de encuentro" idóneo para que se lleve a cabo la reacción.
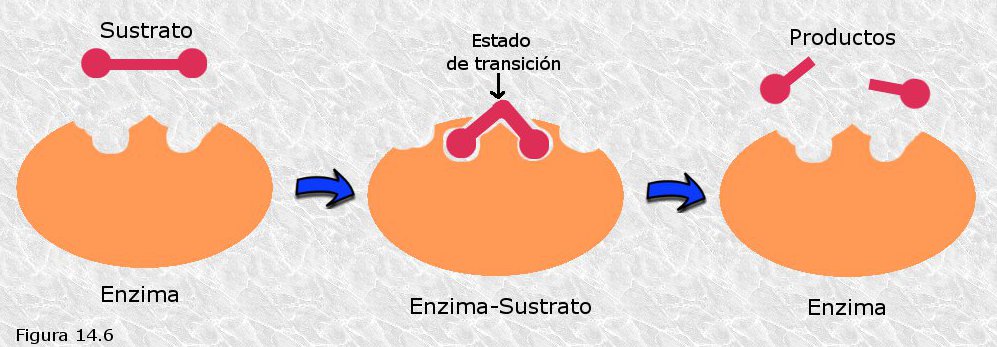
La combinación de los factores de proximidad y orientación favorable de los sustratos sobre el centro activo explica por sí misma los incrementos de velocidad observados en algunas reacciones catalizadas enzimáticamente. Sin embargo, los enzimas todavía pueden utilizar la energía de fijación de un modo adicional para producir catálisis, a saber, provocando en la(s) molécula(s) de sustrato una distorsión favorable para que se alcance con prontitud el estado de transición. ¿Cómo consiguen los enzimas provocar tal distorsión en su(s) sustrato(s)? Para contestar a esta pregunta es necesario revisar algunos conceptos que hasta ahora hemos tratado de una manera quizás excesivamente dogmática. La elegante idea de que dos moléculas con superficies complementarias "encajan" tal y como lo harían "una llave y una cerradura" ha resultado extraordinariamente útil a la hora de explicar muchos procesos bioquímicos; sin embargo, esta idea puede resultar en cierto modo engañosa cuando se aplica a la catálisis enzimática. Un enzima que fuese exactamente complementario con su sustrato sería con toda probabilidad un pésimo enzima, puesto que estabilizaría al sustrato en lugar de inducir su transformación. Dicho de otro modo, el descenso de energía libre que acompañaría a la formación del complejo enzima-sustrato, gracias a la formación de múltiples interacciones débiles entre ambos, sumiría a éste en una especie de "pozo" energético del que le sería muy difícil salir. Este tipo de consideraciones teóricas, apoyadas en muchos casos por resultados experimentales, han conducido a los investigadores de la catálisis enzimática a afirmar que los enzimas no son exactamente complementarios con sus sustratos sino más bien con las especies del estado de transición, es decir, las interacciones débiles que se forman entre el enzima y su sustrato son óptimas en el estado de transición. Cuando el sustrato penetra en el centro activo del enzima se forman algunas interacciones débiles entre ambos; a partir de este momento, cualquier distorsión del sustrato tendente a alcanzar el estado de transición se verá acompañada por el establecimiento de interacciones débiles adicionales energéticamente favorables. Así, el coste energético que supone llevar al sustrato al estado de transición es "pagado" gracias al descenso de energía libre asociado con el establecimiento de dichas interacciones débiles (Figura 14.6). Una vez más, el concepto de energía de fijación resulta crucial para entender cómo los enzimas rebajan la barrera de energía de activación para aumentar la velocidad de una reacción química. Es conveniente resaltar el hecho de que la distorsión del sustrato a la que hemos hecho referencia no es sólo estructural, es decir, no afecta sólo a la forma de la molécula, ángulos o distancias de enlace, sino también electrónica: la formación de interacciones débiles puede provocar cambios en la distribución global de la carga eléctrica, aparición de cargas parciales donde antes no las había, etc.
La energía de fijación del sustrato al centro activo todavía puede suponer una fuente adicional de poder catalítico. La optimización de las interacciones débiles entre el sustrato y el enzima puede conducir no sólo a una distorsión estructural y electrónica del sustrato, sino también a una alteración en la conformación de la proteína enzimática. Tal alteración conformacional, conocida como encaje inducido, puede conllevar una aproximación de grupos catalíticos esenciales del enzima a los grupos funcionales del sustrato susceptibles de reaccionar, con lo que las propiedades catalíticas del enzima se habrán visto incrementadas. La vieja y en exceso rígida imagen de la "llave y la cerradura" para describir la interacción específica entre el sustrato y el centro activo del enzima, ha sido sustituida por una más flexible en la que dicha interacción se contempla más bien como la que tiene lugar entre "una mano y un guante", reflejando así el ajuste mutuo que tiene lugar entre ambos objetos.
El importante papel que juega la energía de fijación en la catálisis enzimática da respuesta, por añadidura, a otra vieja pregunta que se formulaban los primeros investigadores de la naturaleza y acción de los enzimas: ¿por qué los enzimas han de ser tan grandes con respecto a sus sustratos? En efecto: si la energía de fijación es la principal fuente de poder catalítico, el enzima debe ofrecer el mayor número posible de grupos funcionales capaces de establecer interacciones débiles con el sustrato, con el consiguiente aumento en el tamaño de la proteína enzimática.
Hasta aquí hemos podido comprobar que los enzimas utilizan con gran eficacia la energía de fijación como fuente principal de energía libre para producir catálisis. En muchos casos esta energía utilizada tal y como se ha descrito es suficiente para explicar los espectaculares aumentos en las velocidades de reacción que los enzimas son capaces de producir. No obstante, una vez fijado el sustrato, el enzima puede recurrir a formas adicionales de catálisis que no están basadas en la energía de fijación sino en el poder catalítico que poseen en sí mismos determinados grupos funcionales del centro activo. Estos grupos funcionales residen en los que hemos llamado anteriormente aminoácidos catalíticos y es su propia naturaleza química la que les permite funcionar como catalizadores. Aunque son múltiples los mecanismos según los cuales actúan estos grupos catalíticos, en general se pueden encuadrar en alguna de las dos siguientes modalidades:
a) Catálisis covalente.- Algunos enzimas se pueden combinar con el sustrato, a través de un grupo catalítico, para formar un intermediario covalente inestable que se descompone con facilidad para formar los productos.
b) Catálisis ácido-base.- La velocidad de algunas reacciones químicas se ve incrementada por la presencia de ácidos o bases en el medio de reacción. En estos casos, al proporcionar grupos funcionales capaces de actuar como dadores o aceptores de protones (carboxilo, amino, etc.), el enzima puede efectuar una catálisis general ácido-básica.
Los diferentes mecanismos que los enzimas ponen en juego para acelerar las reacciones químicas, unos basados en la energía de fijación , otros en la acción de grupos catalíticos específicos, no son mutuamente excluyentes. Cada enzima particular recurre a una determinada combinación de los diferentes mecanismos estudiados en la que pueden estar presentes todos o tan sólo algunos de ellos; la contribución específica que cada mecanismo aporta al hecho catalítico también es diferente según de qué enzima se trate. Todo parece indicar que, como regla general, los mecanismos basados en la energía de fijación son responsables en mayor medida de los aumentos en la velocidad de reacción que los basados en grupos catalíticos específicos (catálisis covalente y ácido-base)
En la actualidad se han podido estudiar en detalle los mecanismos moleculares responsables de la actividad catalítica de unos cuantos enzimas confirmándose en líneas generales las ideas desarrolladas a lo largo del presente parágrafo.
6.-ESPECIFICIDAD DE LOS ENZIMAS.
Los enzimas, además de ser unos catalizadores muy eficaces,
presentan un alto grado de especificidad química, es decir,
son capaces de inducir la transformación de un sólo tipo de moléculas y
no de otros que también se encuentran presentes en el medio de reacción.
Un enzima es capaz de discriminar entre dos sustancias que
potencialmente podrían actuar como sustratos.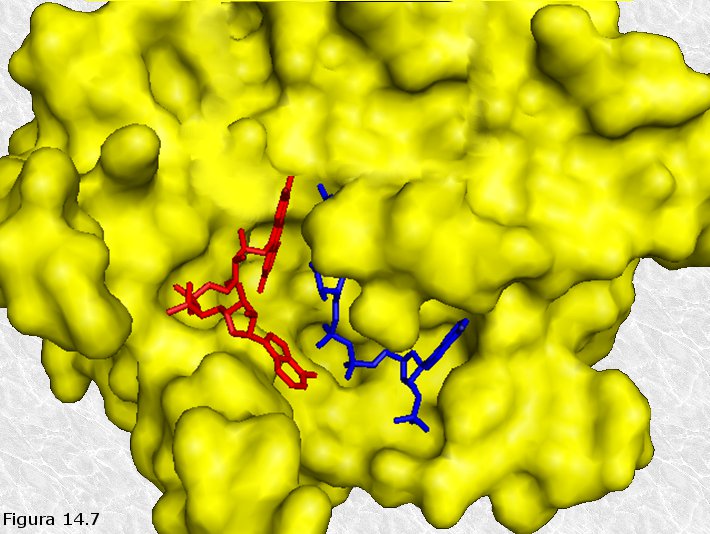
Como ya se apuntó anteriormente, la relación que existe entre el enzima y su sustrato es un caso particular de un fenómeno más amplio: la relación entre las proteínas y sus respectivos ligandos. Aunque hemos introducido alguna salvedad sobre el particular (el enzima no es exactamente complementario con el sustrato sino más bien con el estado de transición), podemos afirmar que entre el enzima y su sustrato se da un acoplamiento espacial (el sustrato "encaja" en el centro activo del enzima) y químico (ambos poseen grupos funcionales que pueden establecer interacciones débiles entre sí). La especificidad de los enzimas reside en esta complementariedad estructural (Figura 14.7). Así, aquellos potenciales sustratos que, por falta de acoplamiento espacial y/o químico, no puedan acceder al centro activo del enzima, no podrán ser transformados por él. Por otra parte, es necesario tener en cuenta que, en muchos casos, no es la totalidad de la molécula de sustrato, sino solamente una parte de ella, denominada grupo determinante de la posición, la que se acopla espacial y químicamente con el centro activo.
La importancia de las interacciones débiles entre grupos funcionales complementarios del sustrato y del centro activo en la determinación de la especificidad de los enzimas debe hacernos reflexionar sobre un hecho crucial: la energía de fijación, que como hemos visto es la principal fuente de energía libre para la catálisis, proporciona también especificidad. Catálisis y especificidad son dos propiedades de los enzimas que, aunque conceptualmente se distinguen con facilidad, responden en realidad a un mismo fenómeno: la interacción energéticamente favorable entre el enzima y el sustrato.
Aunque los enzimas son en general muy específicos comparados con cualquier catalizador artificial, su grado de especificidad resulta ser muy variado. Existen enzimas con una especificidad muy estricta que sólo reconocen a un sustrato determinado y no inducen la transformación de otras moléculas aunque estén estructuralmente muy relacionadas con él; algunos enzimas incluso son capaces de distinguir entre formas estereoisómeras de una misma sustancia (por ejemplo la lactato-deshidrogenasa sólo es capaz de deshidrogenar al estereoisómero L del ácido láctico). En el otro extremo del espectro de especificidades se encuentran enzimas con una especificidad relativamente amplia: pueden inducir la transformación de toda una serie de sustratos que presentan determinado rasgo estructural común (por ejemplo la fosfatasa alcalina induce la hidrólisis de diferentes ésteres del ácido fosfórico). Existen entre ambos extremos todos los grados de especificidad imaginables.
7.-FACTORES QUE AFECTAN A LA ACTIVIDAD ENZIMÁTICA.
Diferentes factores ambientales pueden afectar a la actividad enzimática. Destacaremos dos: el pH y la temperatura.
7.1.-EFECTO DEL pH.
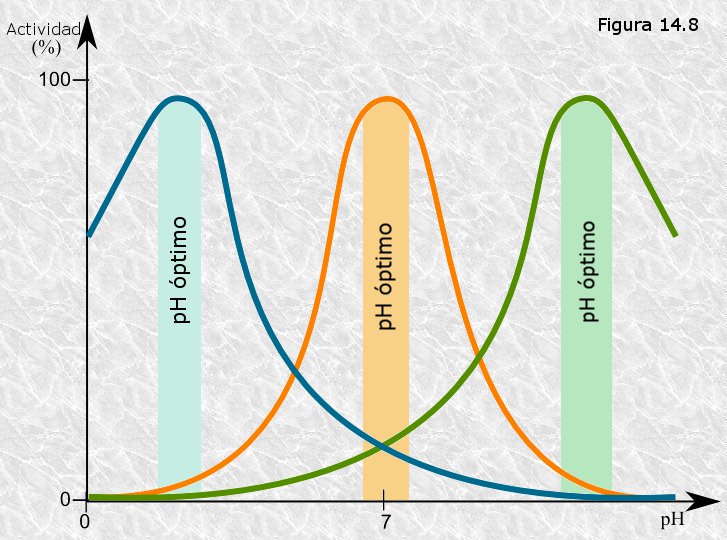 La mayoría de los enzimas presentan un pH óptimo para
el cual su actividad es máxima; por encima o por debajo de ese pH la
actividad disminuye bruscamente. Este efecto se debe a que, al ser los
enzimas de naturaleza proteica, al igual que otras proteínas, se
desnaturalizan y pierden su actividad si el pH varía más allá de unos
límites estrechos (Figura
14.8). De ahí la conocida importancia biológica de los sistemas
tampón.
La mayoría de los enzimas presentan un pH óptimo para
el cual su actividad es máxima; por encima o por debajo de ese pH la
actividad disminuye bruscamente. Este efecto se debe a que, al ser los
enzimas de naturaleza proteica, al igual que otras proteínas, se
desnaturalizan y pierden su actividad si el pH varía más allá de unos
límites estrechos (Figura
14.8). De ahí la conocida importancia biológica de los sistemas
tampón.
En la mayor parte de los casos el pH óptimo está próximo a la neutralidad, en consonancia con el pH intracelular, pero existen enzimas con pH óptimo muy diverso según sea el pH del medio en el que habitualmente actúan (los enzimas proteolíticos del jugo gástrico tienen pHs óptimos próximos a 2 ya que este es el pH de dicho jugo). Por último existen algunos enzimas a los que el pH no afecta en absoluto.
7.2.-EFECTO DE LA TEMPERATURA.
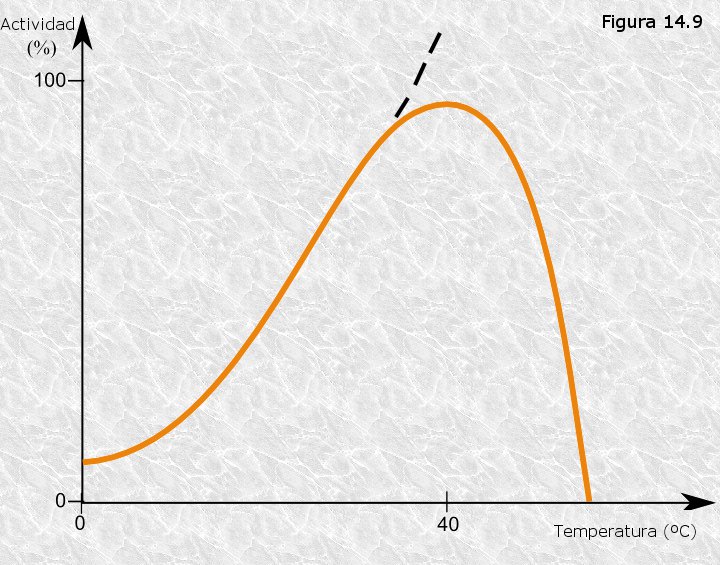 Al igual que ocurre con la mayoría de las reacciones
químicas, la velocidad de las reacciones catalizadas por enzimas se
incrementa con la temperatura. La variación de la actividad enzimática
con la temperatura es diferente de unos enzimas a otros en función de la
barrera de energía de activación de la reacción catalizada. Sin embargo,
a diferencia de lo que ocurre en otras reacciones químicas, en las
reacciones catalizadas por enzimas se produce un brusco descenso de la
actividad cuando se alcanza una temperatura crítica. Este efecto no es
más que un reflejo de la desnaturalización térmica del enzima cuando se
alcanza dicha temperatura. Si representamos gráficamente la variación de
la actividad de los enzimas en función de la temperatura (ver
Figura 14.9) da la impresión de que existe una temperatura "óptima"
análoga al pH óptimo estudiado anteriormente; hay que resaltar que esa
aparente temperatura óptima no es más que el resultado de dos procesos
contrapuestos: 1) el incremento habitual de la velocidad de reacción con
la temperatura y 2) la desnaturalización térmica del enzima.
Al igual que ocurre con la mayoría de las reacciones
químicas, la velocidad de las reacciones catalizadas por enzimas se
incrementa con la temperatura. La variación de la actividad enzimática
con la temperatura es diferente de unos enzimas a otros en función de la
barrera de energía de activación de la reacción catalizada. Sin embargo,
a diferencia de lo que ocurre en otras reacciones químicas, en las
reacciones catalizadas por enzimas se produce un brusco descenso de la
actividad cuando se alcanza una temperatura crítica. Este efecto no es
más que un reflejo de la desnaturalización térmica del enzima cuando se
alcanza dicha temperatura. Si representamos gráficamente la variación de
la actividad de los enzimas en función de la temperatura (ver
Figura 14.9) da la impresión de que existe una temperatura "óptima"
análoga al pH óptimo estudiado anteriormente; hay que resaltar que esa
aparente temperatura óptima no es más que el resultado de dos procesos
contrapuestos: 1) el incremento habitual de la velocidad de reacción con
la temperatura y 2) la desnaturalización térmica del enzima.
8.-NOMENCLATURA Y CLASIFICACIÓN.
Inicialmente se designaba a los enzimas añadiendo el sufijo -asa al nombre del sustrato, o bien a una palabra que describe su actividad. Por ejemplo la ureasa cataliza la hidrólisis de la urea para rendir CO2 y agua, la arginasa cataliza la hidrólisis del aminoácido arginina y la DNA polimerasa cataliza la síntesis del DNA. Sin embargo, a medida que el número de enzimas conocidos iba aumentando, este tipo de nomenclatura comenzó a revelarse poco operativa y en ocasiones ambigua, por lo que se ha adoptado una clasificación sistemática elaborada por una Comisión Internacional de Enzimas reunida a tal efecto. El nuevo sistema divide a los enzimas en seis clases principales, cada una de las cuales se divide a su vez en subclases y éstas en sub-subclases atendiendo al tipo de reacción catalizada. Cada enzima es designada de tres modos: 1) un nombre recomendado, generalmente corto y apropiado para su uso habitual, 2) un nombre sistemático que identifica la reacción que cataliza, y 3) un número de clasificación, que se emplea cuando se precisa una identificación inequívoca del enzima. Veamos como ejemplo el del enzima que cataliza la siguiente reacción.
ATP + CREATINA —› ADP + FOSFOCREATINA
Nombre recomendado: CREATIN-KINASA
Nombre sistemático: ATP:CREATIN FOSFOTRANSFERASA
Número de clasificación: EC 2.7.3.2.
En el número de clasificación EC es la abreviatura de Comisión de Enzimas; el primer dígito (2) indica la clase a la que pertenece el enzima, en este caso la clase transferasas (ver tabla); el segundo dígito (7) indica la subclase (fosfotransferasas); el tercer dígito (3) la sub-subclase (fosfotransferasas con grupo nitrogenado como aceptor); el cuarto dígito (2) identifica inequívocamente al enzima en cuestión.
9.-INHIBICIÓN ENZIMATICA.
Existen una serie de sustancias, llamadas inhibidores, que inhiben o anulan la acción de los enzimas sin ser transformados por ellos. Su estudio resulta de gran utilidad a la hora de comprender los mecanismos de catálisis, la especificidad de los enzimas y otros aspectos de la actividad enzimática.
La inhibición enzimática puede ser irreversible o reversible, esta última comprende a su vez tres tipos: inhibición competitiva, acompetitiva y no competitiva.
9.1.-INHIBICIÓN IRREVERSIBLE.
Algunos inhibidores se combinan de modo permanente con el enzima uniéndose covalentemente a algún grupo funcional esencial para la catálisis con lo que el enzima queda inactivado irreversiblemente. El estudio de este tipo de inhibidores ha resultado de gran utilidad para identificar los grupos funcionales esenciales para la catálisis en aquellos enzimas a los que inactivan.
Este tipo de inhibición se conoce también como "envenenamiento" del enzima. Por ejemplo algunos compuestos organofosforados tóxicos llamados venenos nerviosos, que se utilizan como insecticidas, actúan inhibiendo irreversiblemente al enzima acetilcolinesterasa, la cual interviene en la actividad del sistema nervioso. Se sabe que estos compuestos organofosforados inactivan al enzima formando un enlace éster fosfórico con el grupo hidroxilo de un determinado resto del aminoácido serina, lo que demuestra que ese grupo funcional es esencial para la catálisis.
9.2.-INHIBICIÓN REVERSIBLE.
Los inhibidores reversibles se combinan transitoriamente con el enzima, de manera parecida a como lo hacen los propios sustratos. Algunos inhibidores reversibles no se combinan con el enzima libre sino con el complejo enzima-sustrato.
Se distinguen tres tipos de inhibición reversible:
9.2.1.-INHIBICIÓN COMPETITIVA.
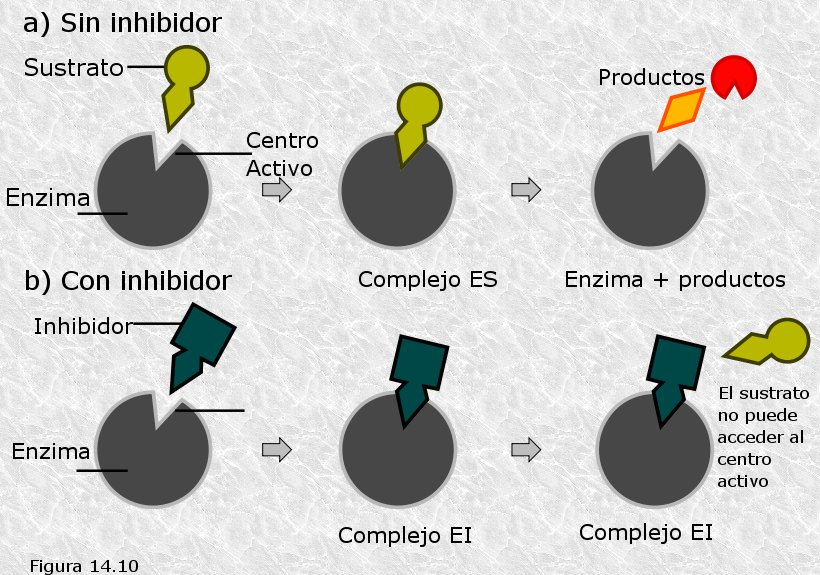
El inhibidor es una molécula que presenta un cierto parecido estructural con el sustrato, de manera que puede competir con él por acceder al centro activo, pero que no posee ningún enlace susceptible de ser atacado por el enzima (Figura 14.10). El inhibidor forma con el enzima libre un complejo enzima-inhibidor de características cinéticas análogas a las del complejo enzima-sustrato, pero que, lógicamente, no puede descomponerse a continuación para dar lugar al enzima libre y a los productos:
9.2.2.-INHIBICIÓN INCOMPETITIVA.
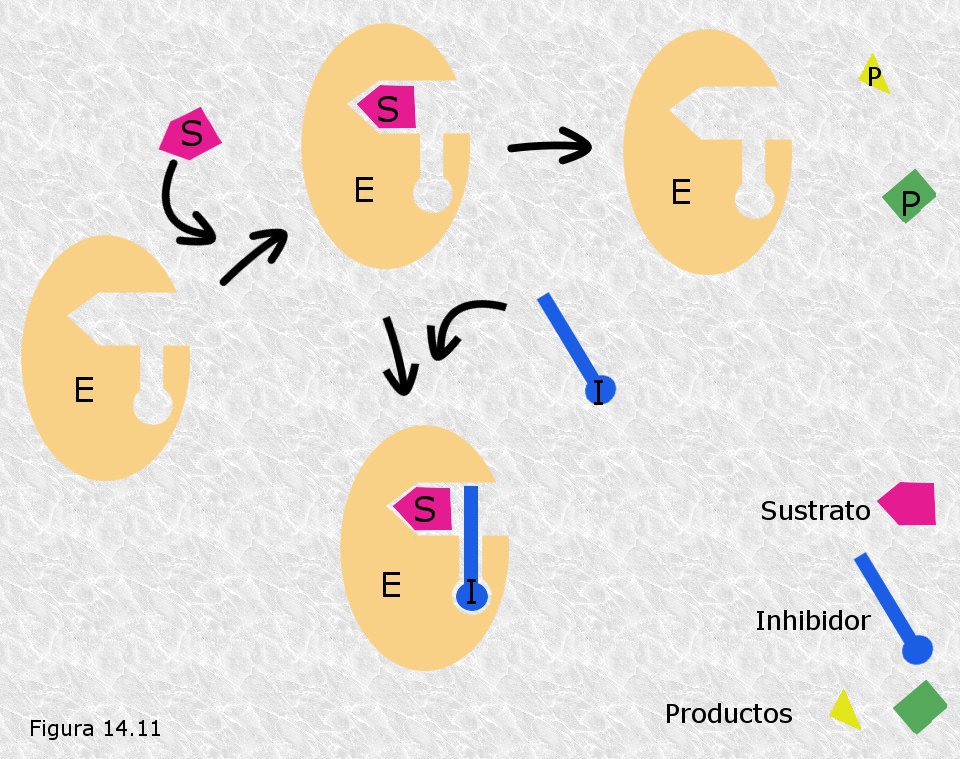
El inhibidor no se combina con el enzima libre ni afecta a su unión al sustrato, sino que lo hace con el complejo enzima-sustrato dando lugar a un complejo inactivo enzima-sustrato-inhibidor, que no se descompone posteriormente para dar lugar a los productos (Figura 14.11). El inhibidor se coloca próximo al centro activo situado de tal manera que impide físicamente la salida de los productos:
9.2.3.-INHIBICIÓN NO COMPETITIVA.
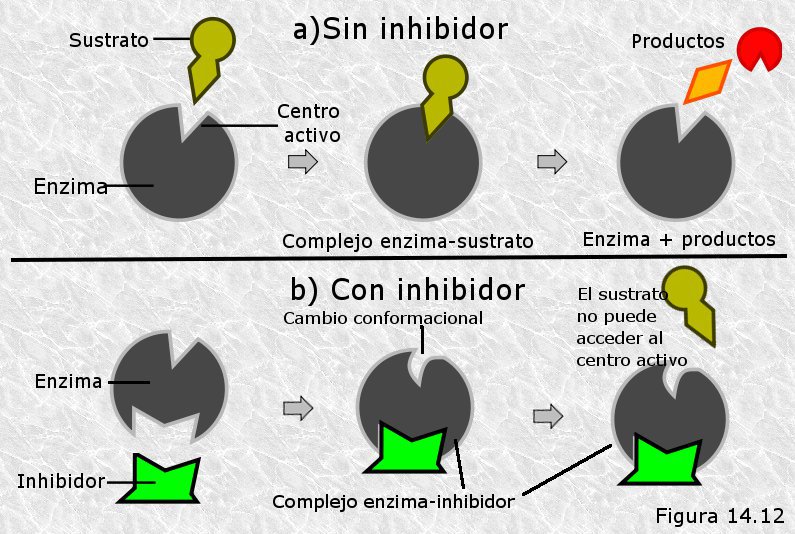
El inhibidor puede combinarse con el enzima libre o bien con el complejo enzima-sustrato, interfiriendo en la acción de ambos. Los inhibidores no competitivos se unen a un lugar del enzima diferente del centro activo provocando en el una alteración que dificulta bien la formación del complejo enzima-sustrato o bien la descomposición de éste para dar lugar a los productos (Figura 14.12). La unión con el inhibidor produce dos formas inactivas: los complejos EI y ESI, ninguna de las cuales puede descomponerse para dar lugar a los productos y al enzima libre:
Aunque podría pensarse que los distintos tipos de inhibición estudiados pueden desempeñar algún papel en la regulación de la actividad enzimática, todo parece indicar que no es así; la regulación de la actividad enzimática se lleva a cabo mediante mecanismos que no se ajustan a ninguno de los modelos de inhibición estudiados y que se describirán en el próximo apartado. El interés del estudio de la inhibición enzimática reside más en su utilidad para comprender la estructura, mecanismos catalíticos y especificidad de los enzimas, que en una importancia biológica real.
10.-ENZIMAS REGULADORES.
La célula es una máquina química que debe ser capaz de autoajustarse o regular su propio funcionamiento para no desperdiciar tiempo ni energía en realizar procesos que no le son útiles en un momento dado, siguiendo así un principio de máxima economía molecular. Este autoajuste se lleva a cabo a varios niveles entre los que destaca la regulación de la propia actividad enzimática.
Todos los enzimas presentan propiedades que los hacen candidatos a constituir elementos reguladores del metabolismo. Estas propiedades son su sensibilidad a los cambios de pH, a la concentración del sustrato o a la concentración de otras sustancias accesorias que denominaremos cofactores. Sin embargo, existen una serie de enzimas que, además de estas propiedades comunes a todos ellos, poseen otras que les confieren un papel específicamente regulador del metabolismo: son los enzimas reguladores. Existen dos tipos principales de enzimas reguladores: los enzimas alostéricos y los enzimas modulados covalentemente. Ambos tipos son responsables de alteraciones en el estado metabólico de las células en intervalos cortos de tiempo (los enzimas alostéricos en cuestión de segundos, los modulados covalentemente en cuestión de minutos).
10.1.-ENZIMAS ALOSTÉRICOS.
Los enzimas alostéricos son aquellos que, además del centro activo mediante el cual interactúan con el sustrato, poseen otro centro de unión llamado centro alostérico mediante el cual interactúan con otra molécula denominada efector o modulador (la palabra "alostérico" hace referencia a la existencia de ese "otro lugar"). La interacción del modulador con el centro alostérico es tan específica como lo es la interacción del sustrato con el centro activo y también está basada en la complementariedad estructural (Figura 14.13). Los enzimas alostéricos presentan pesos moleculares en general superiores a los de otros enzimas y en la mayor parte de los casos son proteínas oligoméricas, es decir están formados por varias subunidades (normalmente en número par).
Los moduladores alostéricos pueden ser de dos tipos: unos estimulan la actividad del enzima al unirse al centro alostérico, reciben el nombre de moduladores positivos o activadores; otros la inhiben y se llaman moduladores negativos o inhibidores. Los inhibidores alostéricos no responden a ninguno de los modelos de inhibición enzimática estudiados en el apartado anterior.
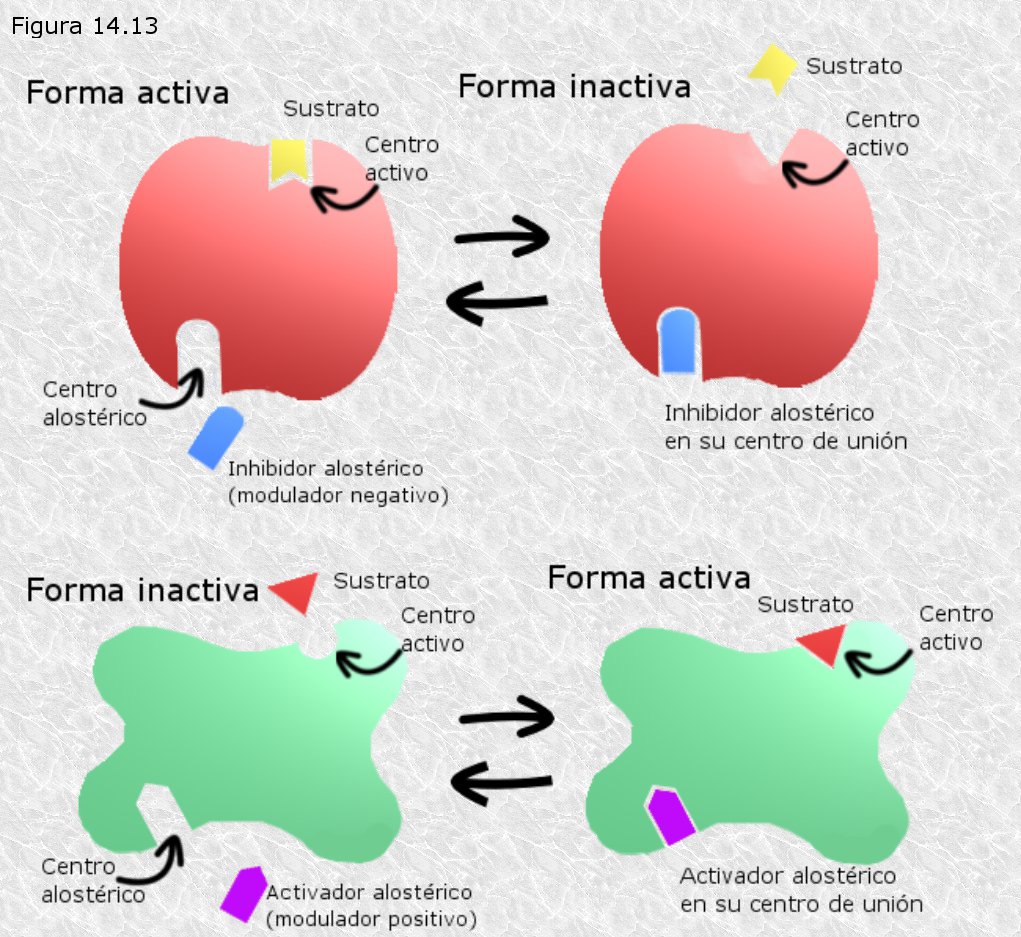
Los enzimas alostéricos presentan siempre dos formas, una activa y otra inactiva, interconvertibles por efecto del modulador (Figura 14.13). Existen dos tipos de control alostérico: el control heterotrópico que se da cuando el modulador es una molécula diferente del sustrato, y el control homotrópico que se da cuando el modulador es el propio sustrato. En ambos casos el modulador puede ser positivo o negativo. Los enzimas con control homotrópico poseen dos o más centros de unión para el sustrato; en ellos la interconversión entre las formas activa e inactiva depende de cuántos sean los centros de unión que estén ocupados por moléculas de sustrato.
Un caso muy común de regulación del metabolismo mediante enzimas alostéricos es la la inhibición por el producto final, también llamada retroinhibición o control feed-back. En ella, el producto final de una ruta metabólica inhibe alostéricamente al enzima que cataliza la primera reacción de dicha ruta, interrumpiendo así su propia síntesis cuando ésta ya no es necesaria. Este tipo de control es muy rentable para la célula, ya que no se interrumpe solamente la síntesis del producto final sino la de todos los intermediarios. Se trata de un control heterotrópico mediante un modulador negativo.
Otro caso es el del sustrato de la primera reacción de una ruta metabólica que actúa como activador del enzima que cataliza dicha reacción. Se trataría aquí de un control homotrópico mediante modulador positivo.
Aunque existen enzimas alostéricos monovalentes, que responden a un sólo modulador, la inmensa mayoría son enzimas polivalentes, que poseen varios centros alostéricos mediante los cuales interactúan con distintos moduladores positivos y/o negativos, presentando un tipo de control mixto homotrópico-heterotrópico.
10.2.-ENZIMAS MODULADOS COVALENTEMENTE.
Los enzimas modulados covalentemente también presentan dos formas, una activa y otra inactiva, que son interconvertibles por modificación covalente de sus estructuras catalizada por otros enzimas, denominados enzimas moduladores. Tal modificación suele consistir en la adición o eliminación de un grupo químico esencial para la catálisis (generalmente un grupo fosfato, metilo, adenilato u otros) de manera que el enzima modulado se activa cuando está unido a dicho grupo y se inactiva cuando éste se elimina. En la mayor parte de los casos son necesarios dos enzimas moduladores diferentes, uno que activa el enzima modulado y otro que lo inactiva. Un ejemplo clásico de modulación covalente lo constituye el enzima glucógeno-fosforilasa, que actúa en el metabolismo de los glúcidos liberando unidades de glucosa-1-fosfato a partir de las cadenas polisacarídicas del glucógeno. La glucógeno-fosforilasa es activada por un enzima modulador, la fosforilasa-quinasa, que une covalentemente un grupo fosfato a un resto específico de serina en cada una de las dos subunidades del enzima modulado. Otro enzima modulador, la fosforilasa-fosfatasa, escinde hidrolíticamente dichos grupos fosfato desactivando así el enzima (Figura 14.14).
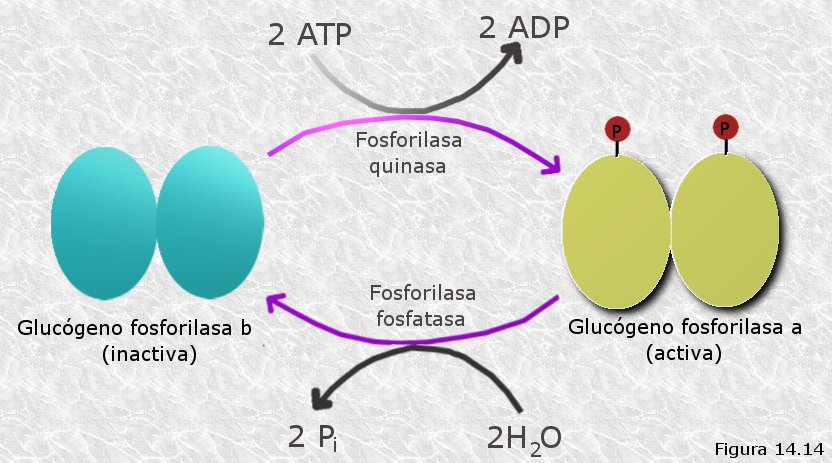
La modulación covalente tiene la ventaja de que puede utilizarse para amplificar una señal química, ya que una sola molécula del enzima modulador puede activar o desactivar a muchas moléculas del enzima modulado, que a su vez podrán actuar o no sobre un elevado número de moléculas de sustrato, produciéndose así lo que se conoce como efecto cascada. En muchos casos, los enzimas moduladores a su vez pueden activarse o desactivarse como respuesta a una señal hormonal. Por ejemplo, la hormona denominada adrenalina actúa sobre receptores específicos de la membrana de las células musculares desencadenando un proceso en el que están implicados varios enzimas moduladores que a su vez resultan modulados covalentemente por otros enzimas moduladores de un nivel superior. El resultado final de dicho proceso es una degradación masiva de glucógeno a glucosa-1-fosfato destinada a la producción de energía para las células musculares. De este modo, una débil señal química, consistente en unas pocas moléculas de hormona, es amplificada en varios escalones para producir un efecto metabólico de grandes proporciones.
Un caso particular de este tipo de regulación es la la activación covalente de los zimógenos. Algunos enzimas se sintetizan dentro de las células en formas inactivas denominadas zimógenos. Una vez secretados al exterior de la célula son activados mediante la escisión hidrolítica catalizada enzimáticamente de algunos péptidos de su cadena polipeptídica. Este tipo de activación es irreversible. El ejemplo clásico de este tipo de regulación es el de los enzimas digestivos pepsina, tripsina y quimotripsina que, con el objeto de que evitar que lleven a cabo su actividad degradativa en el interior de las células, se sintetizan en forma de sus respectivos zimógenos inactivos y sólo se activan una vez han sido secretados al tracto digestivo.
11.-COFACTORES ENZIMÁTICOS.
Algunos enzimas necesitan para llevar a cabo su actividad catalítica de la concurrencia de una o más sustancias de naturaleza no proteica que reciben el nombre de cofactores. No debe entenderse que los cofactores son sustancias que meramente potencian la actividad enzimática sino que, en determinados enzimas, son absolutamente imprescindibles para que ésta se realice. En ausencia de cofactor el enzima resulta catalíticamente inactivo y recibe el nombre de apoenzima; la combinación de apoenzima y cofactor da el holoenzima catalíticamente activo. Existen dos tipos de cofactores enzimáticos: los iones metálicos y los coenzimas.
Los iones metálicos que actúan como cofactores enzimáticos son generalmente cationes mono o divalentes. Pueden actuar de varias maneras: 1) En algunos enzimas el ion metálico constituye el verdadero centro catalítico; en estos casos el ion suele presentar por sí solo una cierta actividad catalítica, que se ve incrementada cuando forma parte del enzima. 2) En otros enzimas el ion metálico constituye un grupo puente para unir el sustrato al centro activo. 3) A veces el ion metálico no forma parte del centro activo sino que se encuentra en un lugar del enzima muy alejado del mismo, actuando como agente estabilizador de la conformación nativa del enzima.
Cuando el cofactor es una sustancia orgánica de naturaleza no proteica recibe el nombre de coenzima. Los coenzimas actúan generalmente como transportadores intermediarios de grupos funcionales, de determinados átomos o de electrones, los cuales son transferidos de una sustancia a otra en la reacción enzimática global. A veces los coenzimas se hallan íntimamente unidos a la molécula proteica constituyendo un verdadero grupo prostético. En otros casos la unión es débil y el coenzima actúa en realidad como si de un sustrato más del enzima se tratase.
Cuando se analiza la estructura química de muchos coenzimas se comprueba que tienen formando parte de ella a alguna de las sustancias conocidas como vitaminas. Existe pues una clara relación entre vitaminas y coenzimas.
12.- VITAMINAS.
Las vitaminas son una serie de sustancias de naturaleza química variada que, en cantidades mínimas, son necesarias para el normal desarrollo y funcionamiento de muchos organismos. Su importancia biológica se manifestó debido a que algunos organismos no las pueden sintetizar y deben adquirirlas por tanto de procedencia exógena.
Desde muy antiguo se sabía que determinados alimentos tenían propiedades curativas o preventivas para determinadas enfermedades. El fraccionamiento químico de estos alimentos condujo al aislamiento y purificación de las sustancias responsables de este efecto preventivo, las cuales recibieron el nombre de vitaminas debido a que la primera que se identificó era una amina (de "vital amina"). Pudo constatarse entonces su variada naturaleza química, siendo clasificadas en hidrosolubles y liposolubles según su mayor o menor afinidad por el agua o por las sustancias lipídicas.
En la actualidad está perfectamente establecida la función coenzimática de la mayoría de las vitaminas hidrosolubles: a excepción de la vitamina C todas forman parte de alguno de los coenzimas conocidos. Es conveniente resaltar que, aunque existe una clara relación entre las vitaminas hidrosolubles y los diferentes coenzimas que actúan en el metabolismo, vitaminas y coenzimas no son exactamente la misma cosa; un ejemplo nos ayudará a aclararlo: el ácido nicotínico es una vitamina hidrosoluble que no puede ser sintetizada por las células humanas. Ahora bien, dichas células sí pueden transformar el ácido nicotínico en nicotinamida, la cual, unida a otros componentes celulares, forma parte de los coenzimas NAD y NADP, que actúan en el metabolismo como transportadores de electrones. Por otra parte, las vitaminas liposolubles parecen presentar funciones de naturaleza hormonal o reguladora del metabolismo que en algunos casos aún no son bien comprendidas.
Las necesidades exógenas de vitaminas difieren ampliamente de unas especies a otras según éstas sean capaces o no de sintetizarlas. Por ejemplo el hombre necesita 14 vitaminas de procedencia exógena, mientras que la especie bacteriana Escherichia coli sólo necesita una (la biotina) pudiendo sintetizar todas las demás. Por otra parte, sólo los animales superiores parecen necesitar vitaminas liposolubles de procedencia exógena.